El síndrome de Gorlin-Goltz (SGG) fue descrito en 1960 por los autores a los que debe su nombre, Gorlin y Goltz. Inicialmente describieron la triada que caracteriza al síndrome: carcinomas basocelulares múltiples, queratoquistes maxilares y costillas bífidas. También conocido como síndrome névico-basocelular o síndrome del carcinoma nevoide basocelular, entre otros, hoy se sabe que es una enfermedad genética con herencia autosómica dominante. Está causada por la mutación del gen Patched (PTCH), un gen supresor del tumor localizado en el cromosoma 9 (9q22, 3-q31). Su prevalencia estimada varía entre 1:57.000 y 1:164.000 dependiendo del país, estando generalmente aceptada una prevalencia media aproximada de 1:60.0004.

El SGG es una enfermedad caracterizada clínicamente por la predisposición al desarrollo de múltiples neoplasias, así como por la aparición de anomalías del desarrollo. El inicio de la clínica se produce en la infancia o adolescencia y se acompaña de la asociación de un conjunto de manifestaciones, siendo la más común la presencia de numerosos carcinomas basocelulares y queratoquistes odontogénicos maxilares. Otras manifestaciones incluyen: hiperqueratosis palmoplantar, anomalías esqueléticas, calcificaciones intracraneales ectópicas y dismorfia facial (macrocefalia, fisura labiopalatina y anomalías oculares graves). El déficit intelectual está presente en cerca del 5% de los casos. Pueden presentarse también problemas oculares, genitourinarios y cardiovasculares. Un 5-10% de los pacientes con SGG desarrollan meduloblastomas malignos que pueden ser causa potencial de muerte temprana. Se establece su diagnóstico cuando se cumplen 2 criterios clínicos mayores o bien uno mayor y 2 menores (tabla 1). La prueba diagnóstica definitiva es demostrar una mutación en el gen PTCH.
El presente reporte tiene por objetivo mostrar mediante un caso clínico y radiográfico, la importancia de conocer los criterios mayores y menores para el diagnóstico del Síndrome de Gorlin Goltz.

Tabla 1: Criterios mayores y menores para el diagnóstico de SGG. Se establece su diagnóstico cuando se cumplen 2 criterios clínicos mayores o bien uno mayor y 2 menores.
Fig 1. Paciente masculino de 16 años, presenta asimetría facial, prognatismo mandibular, hipertelorismo y estrabismo del ojo izquierdo.
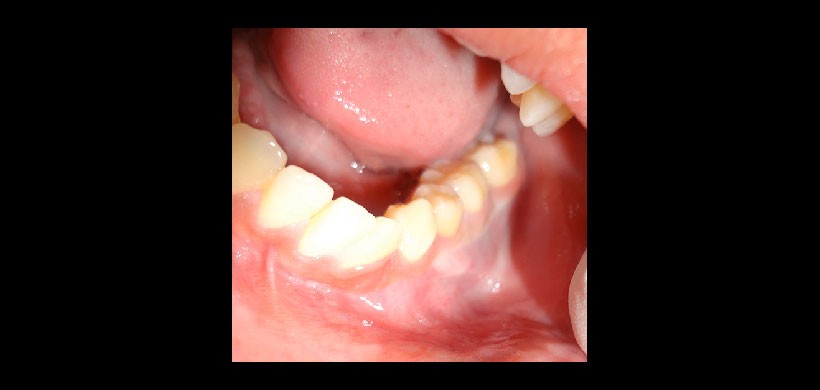
Fig 2. Examen clínico intraoral que evidencia leve aumento de volumen de la tabla ósea vestibular, de consistencia firme.
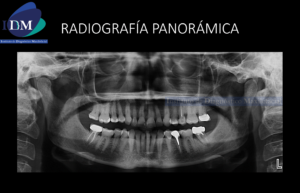
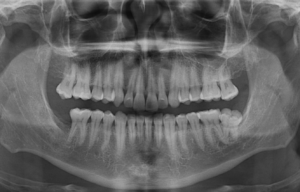
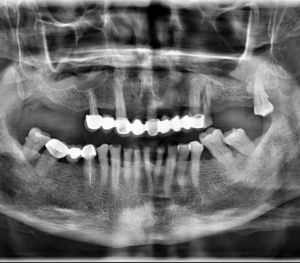
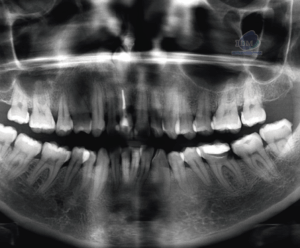
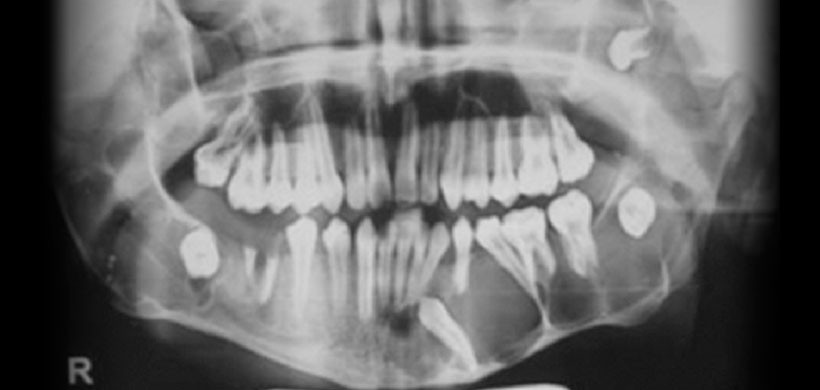
Fig 3. Radiografía panorámica, muestra múltiples lesiones radiolucidas de limites definidos y bordes corticalizados, localizado en cuerpo, y rama mandibular bilateral y region posterior del maxilar superior.
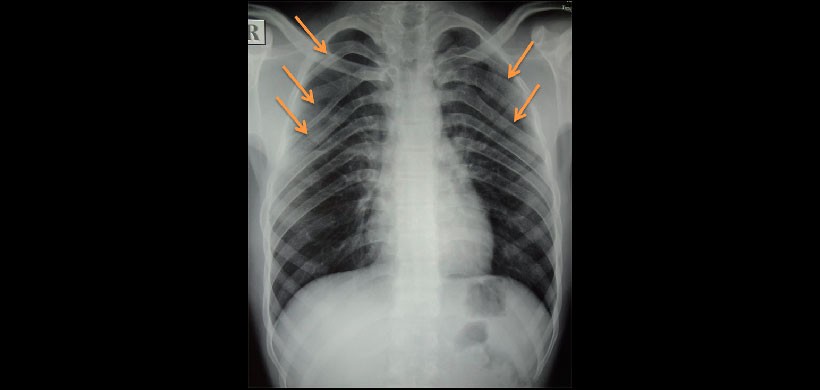
Fig 4. Radiografía de tórax, muestra la presencia de costillas bífidas.
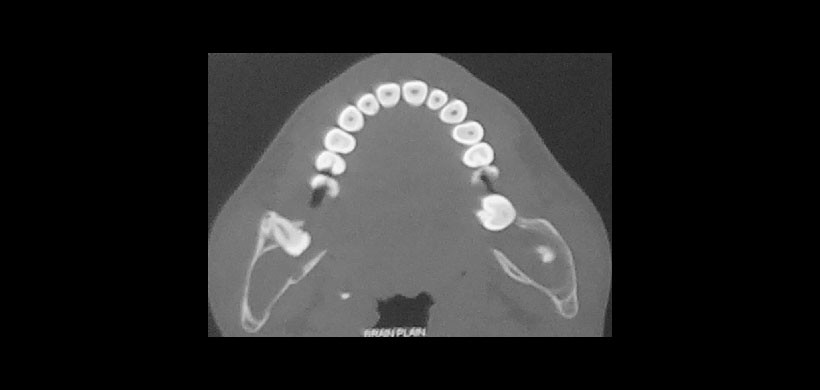
Fig 5. Tomografía de haz cónico, evidencia lesiones osteolíticas asociados a piezas dentarias retenidas produciendo leve expansión de las tablas óseas.
Referencia:
- Manjima S, Naik Z, Keluskar V, Bagewadi A. Multiple jaw cysts-unveiling the Gorlin-Goltz syndrome. Contemp Clin Dent 2015;6:S102-5.
- Gílabert R, Infante P, Redondo P, Torres E, García-Perla A, Cicilia D. Síndrome de Gorlin-Goltz: manejo del carcinoma basocelular facial. rev esp cir oral maxilofac . 2013;35(1):23–30.
Elaborado por : C.D. Luis Alberto Cueva Principe, Mg. Esp. C.D. Andrés Agurto Huerta.